| Recherche en Bioinformatique - Alignement |
| Alignement par paires (Pairwise Sequence Alignment) |
| Définition - Problčme d'alignement par paires |
| Soient deux séquences S1 et S2, une matrice de score w. Le problčme d'alignement par paires consiste ŕ déterminer un alignment de coűt optimal selon w. |
 Enumération exhaustive
Enumération exhaustive
Ainsi que nous l'avons vu précédemment, on cherche ŕ obtenir un alignement de coűt optimal. La méthode de recherche la plus simple et qui vient tout de suite ŕ l'esprit consiste ŕ réaliser une énumération de tous les alignments possibles et ŕ retenir ceux de coűt optimal. Malheureusement le nombre d'alignements différents de deux séquences de męme longueur n est donné par la formule :
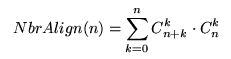
Programmation dynamique
La Programmation Dynamique est la méthode la plus intéressante pour l'alignement de deux séquences car sa complexité temporelle et spatiale est en O(n2).
La Programmation Dynamique est appliquée ŕ des problčmes d'optimisation pour lesquels un choix doit ętre fait entre plusieurs solutions possibles afin d'aboutir ŕ une solution optimale. Le terme Programmation fait ici référence ŕ une méthode basée sur le calcul de tableaux de valeurs.
Exemple d'application 
| L'algorithme d'alignement global basé sur la programmation dynamique a été mis au point par Needleman et Wunsch en 1970 [1]. Une version pour l'alignement local fut ensuite introduite par Smith et Waterman en 1981 [2]. |
Soient deux séquences S = { x1, x2, ... , xN } et T = { y1, y2, ... , yM } ŕ aligner. L'algorithme d'alignement utilise deux matrices de dimensions (N+1) x (M+1) :
Remarque : la matrice des directions n'est pas essentielle car elle peut ętre déduite de la matrice des scores optimaux d'alignement, mais pour des raisons de simplicité d'écriture des algorithmes on utilise généralement cette matrice.
Soient deux indices i, j tels que i,j >= 1. Supposons que nous ayons aligné les préfixes suivants par rapport ŕ une fonction de score w :
| M[i-1][j-1] | M[i-1,j] | |
 |  |
|
| M[i,j-1] |  | M[i,j] |
| Alignement par Paires par Programmation Dynamique | |||||||||||||||||||||||||||
Soient deux séquences S et T de longueurs respectives N et P. La recherche d'un alignement global optimal entre S et T suivant une fonction de score w est obtenu par construction d'une matrice des scores optimaux d'alignement M[0..N,0..P] telle que :
La matrice des directions est obtenue par les formules suivantes :
|
Une fois les matrices M et Dir obtenues, il suffit pour trouver un alignment de parcourir le chemin inverse depuis M[N,P] en remontant vers M[0,0] en se déplaçant dans l'une des trois directions possibles (Nord, Ouest ou Nord-Ouest) uniquement si le mouvement est autorisé par la matrice des directions.
On obtient donc non pas un alignement unique mais un ensemble d'alignements possibles. On peut se limiter ŕ un seul alignement en ne choisissant qu'une direction ŕ chaque étape et en privilégiant une direction en particulier.
Exemple d'application
Remarque finale : le problčme d'alignement est un problčme difficile car on ne sait pas définir en terme qualitatif ce qu'est un bon alignement. L'alignement concerne la séquence primaire or pour juger de la qualité d'un alignement il faudrait pouvoir disposer d'informations supplémentaires concernant la structure secondaire (voire tertiaire) des séquences afin d'aligner des structures de męme type (on peut penser qu'il est plus judicieux d'aligner une hélice alpha avec une autre hélice alpha et non pas avec un feuillet béta).
|
Jean-Michel Richer, 2004